子科生物(https://show.guidechem.com/zikerbio/)报道:泛基因组有助于充分挖掘目标物种的遗传变异资源。2021年5月28日,以四川农业大学为首的研究团队在Cell杂志发表了题为“Pan-genome analysis based on 33 high-quality assemblies provides insights into hidden genomic variations in rice”的研究论文,揭示了水稻基因组中的"隐藏"变异。
结构变异(Structural variation,SVs)和基因拷贝数变异(gene copy number variations,gCNVs)作为重要的遗传变异来源,对于作物进化、驯化和改良具有重要贡献。水稻是世界上最重要的粮食作物之一,也是植物研究中单子叶植物和农作物的主要模式物种。但目前为止,还未有对其SV和gCNV全面准确的研究报道。本研究作者选取33份(32份亚洲栽培稻和1份非洲栽培稻)具有高度代表性的水稻材料,利用平均测序深度为60X的PacBio三代长读长测序结合Bionano等技术组装了31个高质量的水稻基因组,并结合已报道的日本晴和蜀恢R498的基因组,构建了水稻泛基因组和图形基因组。通过系统的基因组比较分析,共鉴定到171,072个SVs和22,549个gCNVs。进一步探究了SV在亚洲栽培稻群体中的分布规律和形成机制,揭示了大量SVs和 gCNVs与基因表达量变化显著相关,实例分析表明SV和gCNV对调控水稻农艺性状的重要作用。首次构建了水稻图形基因组,阐明了在研究农艺性状多样性变异方面,图形基因组和SV具有SNP与线性参考基因组无法代替的作用,搭建了便于使用基因组序列和查询遗传变异等内容的网站。

对选取的遗传背景具有高度代表性的33个水稻材料中的31份材料进行了长片段测序、高质量基因组组装及基因注释。基因组质量评估结果表明所有31个基因组都达到了参考基因组水平。基因组组装中平均鉴定出40,082个蛋白质编码基因,进一步通过BUSCO评估验证了基因注释的高度完整性。以日本晴的基因为基础采用迭代策略构建泛基因组,获得了包含66,636个蛋白质编码基因的栽培稻泛基因组,其中20,374个基因为核心基因,46,262个为可变基因(其中14,609个为单个基因组的私有基因)。组装基因组中平均鉴定出468,144个转座元件(TEs),每个组装基因组中总TEs的平均长度为202.8 Mbp(165.7-211.8 Mbp),平均占组装序列总长度的52.7%。
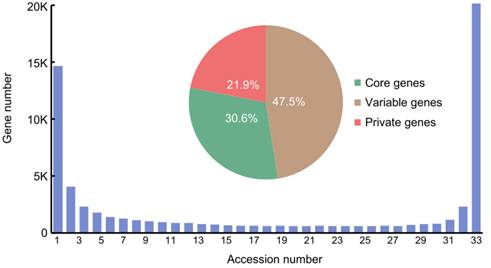
图1:泛基因组基因在 33 个种质中的分布。核心基因存在于所有种质中,可变基因存在于1个以上33个以下的种质中。
二、SVs和SV热点区域的发现
对33个高质量水稻基因组序列的系统比较分析,相对于日本晴基因组,共鉴定到 171,072个非冗余结构性变异(SVs),包括164,009个存在/缺失变异(PAVs),6,109个易位和954个倒位。应用Bionano,Hi-C和PacBio reads等方法证明了其鉴定到SV的高度准确性,通过与已报道的SV比较分析,发现其中绝大多数SV在先前研究中未鉴定到。通过对SV在基因组上的分布以及相对应基因结构分布的分析,发现SV在染色体上呈不均匀分布,共检测到140个SV热点区域,且发现多数SV位于基因的非编码区。在染色体11上一个SV热点区域与12个稻瘟病抗性数量性状基因座(QTLs)重叠或邻接。这些发现与先前报道同时表明位于SV热点区域内的变异可能经历更强的环境选择(图2)。
三、代表亚洲栽培稻衍生状态的 SVs 的推断和表征
利用非洲栽培稻CG14作为外群,对亚洲栽培稻群体中的SV序列的祖先型(derived-state SVs,dSVs )进行了推断,共检测出130,862个dSVs,包括125,889个PAVs、627个倒位和4,346个易位。大多数 dSVs 存在于一个或仅几个种质中。dSVs 的推断有助于理解自然选择和/或人工选择作用于SVs 的方向和生物学意义。例如,先前报道的两个与独脚金内酯合成相关基因SLB1和SLB2被认为是在籼稻群体中缺失导致优良性状(分蘖增加和独脚金合成减少)而被人工选择到,本研究结合该SV的分化状态及群体分布等分析,发现很可能是粳稻中获得SLB1和SLB2基因序列,其原因可能是其能帮助磷的吸收提高产量而被保留下来(图2)。
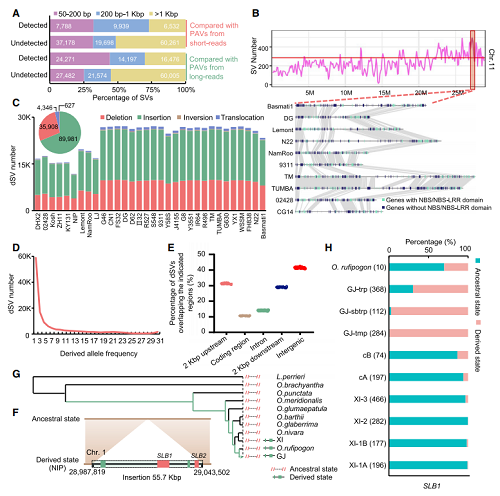
图2:代表派生状态的 SVs 的推理和表征。
四、SVs 具有广泛的基因表达谱
据报道,SVs 通过改变基因序列或影响调控序列来影响附近基因的表达,该研究的数据表明大多数 dSVs 与非编码区域重叠。对蜀恢R527的29种不同样本类型的RNA测序分析发现胁迫处理后dSVs基因表达变化大于3倍的比例明显高于非dSVs基因,表明dSVs基因通常对环境胁迫更敏感。为了研究 dSVs 对特定基因表达的潜在影响,进一步对33份材料进行根和茎的RNA-seq分析,发现15.6%(3,340)的SVs(存在于> 3份但< 29份材料中)与一个基因区域有重叠,这与它们在根和/或芽中相应基因的表达改变有关。这些分析共同表明在水稻进化和驯化过程中SVs具有广泛的基因表达谱。
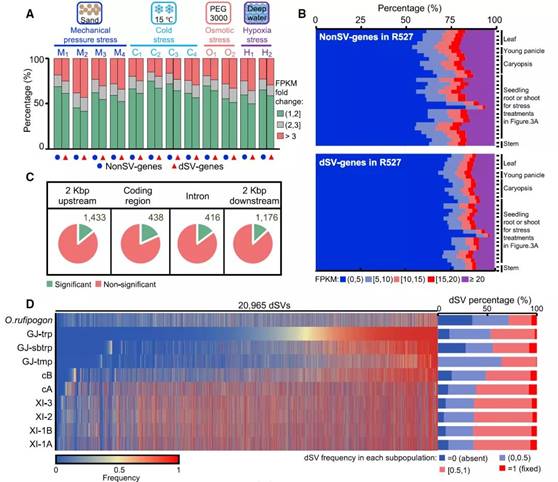
图3:SVs对基因的影响塑造了环境适应和驯化。
五、gCNVs普遍存在并与农艺性状的变异有关
通过研究分析33个高质量基因组序列和泛基因组中基因的蛋白序列数据,25,549个(38.34%)蛋白质编码基因被推断为gCNVs,包括14,782个基因PAVs。其中 2,945 个推断为 CG14 特异性 gCNVs,22,604 个推断为亚洲稻特异性 gCNVs,明显高于先前在水稻基因组规模研究中报告的水平(1,321 个gCNVs)。
这些gCNVs包括先前报道的GL7和Sc的两个拷贝以及320个未报道的功能性研究基因,例如稻瘟病抗性基因(Pid4)和耐旱基因(OsDT11)。296和361个基因的表达水平分别与茎和根中的CNV相关。此外,gCNVs可以导致异位表达模式。总体而言,这个gCNVs目录有助于研究水稻表型变异背后潜在的隐藏基因组变异。
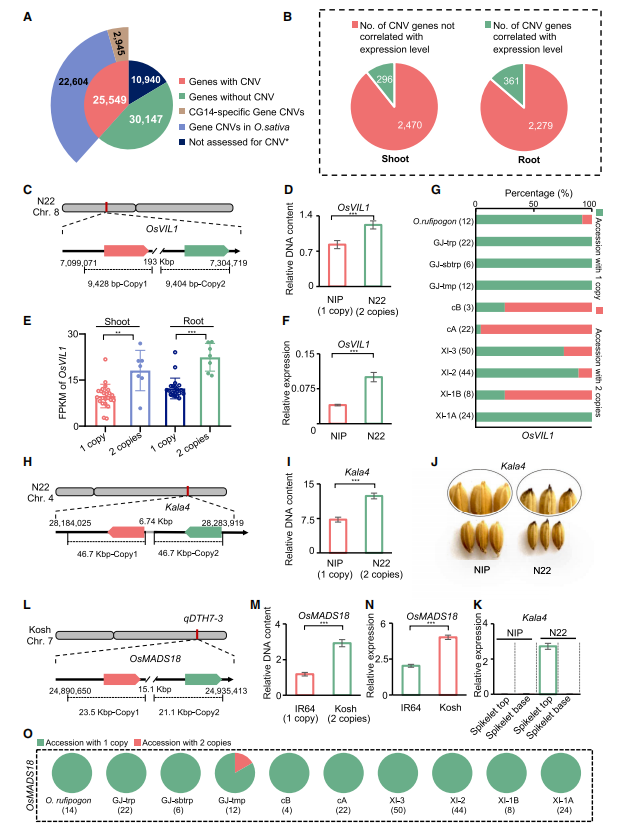
图4:普遍存在的gCNVs与农艺性状变异相关。
六、多种机制驱动水稻中的SV形成
为了更好地了解促成 SV 形成的驱动力和特定机制,作者采用了基于亚洲稻种质中 dSVs 断点序列表征的工作流程分析。发现这些SVs主要的形成机制是转座因子插入(TEI)(50.8%),其次是非同源末端连接(NHEJ)(43.9%)。进一步分析表明2,741个dSVs是通过叉停滞和模板转换/微同源性介导的断裂诱导复制(FoSTeS/MMBIR)机制形成的。由TEI形成的水稻SVs中55%和38.3%分别来自DNA TEs和长末端串联重复(LTRs)。总体而言,研究结果表明TEs,尤其是LTRs,能够以某种方式频繁地为NHEJ产生DNA断裂或通过提供同源序列来促进非等位同源重组(NAHR)。
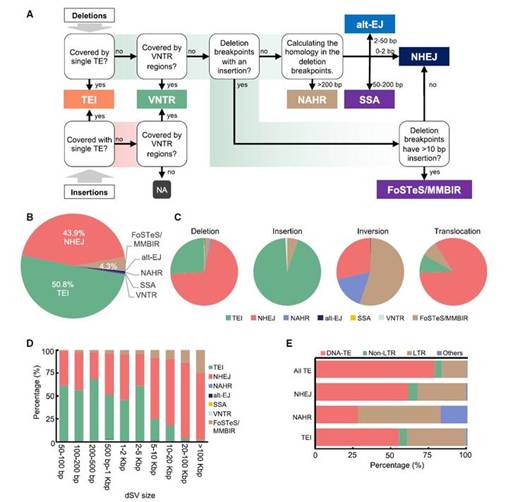
图5:不同机制驱动水稻中 SVs 的形成。
本研究中作者通过对水稻泛基因组的研究,克服了基于短读长测序数据识别 SV 的困难和不可靠性,成功的揭示了水稻中隐藏的SVs和gCNVs。进一步分析了 SV 分布并评估了 SV 形成的机制和 SV 对基因表达的影响,提供了 SVs 和 gCNVs 如何直接影响环境适应性和农艺性状的示例。现今,泛基因组从微生物拓展到植物、动物研究领域,逐渐成为基因组学研究的新热点、新趋势,同时也是PacBio HiFi测序的一大应用热点。
HiFi Reads是PacBio测序平台推出的兼顾长读长和高准确度的测序技术。作为PacBio最新的数据类型,既兼顾读长(15-25kb)又具有高准确度(>99.9%,即Q30准确率)的HiFi reads,不仅可以改善变异检测,减少组装时间,并能够识别复杂的基因组区域的细微差别,有助于增加基因组组装的连续性,准确性和完整性,可以实现多倍体基因分型。
在泛基因组研究中,PacBio HiFi测序可以提供高质量的参考基因组,同时可以有效节约生信分析的时间,大大加速大型泛基因组研究的效率。相信在不久的将来,PacBio HiFi测序将为更为广泛的泛基因组研究带来重要变革。
参考文献:
Qin P , Lu H , Du H , et al. Pan-genome analysis of 33 genetically diverse rice accessions reveals hidden genomic variations[J]. Cell, 2021.